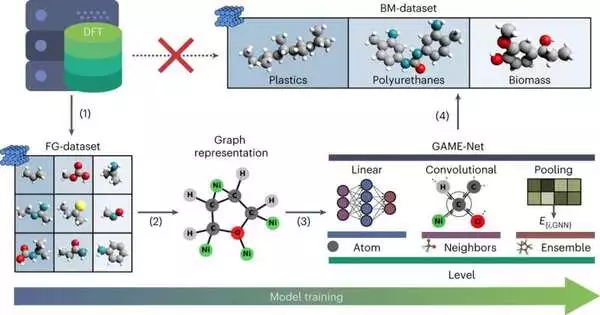
Instead of oil, it will be necessary to use feedstocks like plastics and biomass to achieve sustainability goals. In addition, industries are constantly looking for processes that are both more sustainable and efficient, which frequently necessitates precise simulations. Using density functional theory (DFT), it is essential to estimate catalyst performance in heterogeneous catalysis by determining the energy of molecules adsorbed on surfaces. However, this is not possible for large organic molecules like those found in biomass and plastics.
To resolve this issue, researchers from the Prof. López research bunch at the Organization of Compound Exploration of Catalonia (ICIQ-CERCA) as a team with specialists from The Matter Lab (College of Toronto) have made another model called GAME-Net, a chart brain network that quickly assesses adsorption energy when particles connect to surfaces. The results of their study have been published in Nature Computational Science.
GAME-Net is six orders of magnitude quicker than DFT and trained on a diverse dataset of small and medium-sized molecules containing functional groups. On the test set, the mean absolute error (MAE) is 0.17 eV. The model predicts adsorption energies with a MAE of 0.016 eV/atom when applied to biomass (wood) and plastic molecules with up to 30 atoms.
“Our model demonstrates that AIs are capable of describing complex chemical interactions using extremely simple models, making them more accessible to scientists who want to accelerate their research,”
Dr. Pablo-García and Santiago Morandi.
Using a straightforward molecular representation, GAME-Net can estimate adsorption energy with an accuracy comparable to that of DFT. This ground-breaking model makes it possible to predict the adsorption energy of larger molecules derived from biomass, polyurethanes, and plastics. This makes it possible to study chemical systems that are difficult to model using DFT. Because it can be operated with ease on a laptop, this revolutionary technology is accessible to scientists and researchers worldwide.
“For us, this work is a starting point for putting machine learning algorithms for quantum chemistry into the typical workflow of experimental and computational heterogeneous catalysis researchers.” Even though GAME-Net has some limitations, “this model is a valuable tool for the community because of its speed, simplicity in its structure, and the way it can be interconnected,” says Santiago Morandi from ICIQ.
How about we ‘KISS’: Keep it small and straightforward. The rising popularity of artificial intelligence has prompted numerous efforts to enhance its predictive abilities. Among them are diagram brain organizations (GNNs), a kind of brain network fit for learning the “shape” of information.
The researchers used a straightforward graph representation in their algorithm, with edges representing chemical bonds and nodes representing atoms. A graph neural network is used to predict the change in energy that occurs when a catalyst interacts with a reactant molecule, whereas this change is typically obtained from DFT.
GAME-Net was trained on a wide range of small molecules with various heteroatoms like sulfur, nitrogen, and oxygen. To start the cycle, analysts made the “utilitarian gatherings” (FG) dataset without any preparation, which includes 207 natural atoms adsorbed on 14 change metals (Ag, Au, Cd, Co, Cu, Fe, Ir, Ni, operating system, Pd, Pt, Rh, Ru, and Zn) on their least surface energy aspects.
To guarantee high reproducibility guidelines, every piece of computational information created in this study is available from the ioChem-BD vault. The pre-trained model and GAME-Net’s code are open-source and available for download from a repository under the MIT license.
“With this work, we wanted to see if we could teach an AI complex chemical interactions using high school students’ chemical representations. Dr. Pablo-Garc, a Ph.D. student at ICIQ who is currently enrolled at the University of Toronto, reports that “surprisingly, our AI turned out to be an excellent student.”
This cutting-edge AI framework makes it possible to quickly screen catalytic materials, which is especially useful for systems that are too difficult to simulate using conventional methods. The chemical industry can benefit from incorporating AI into their research to speed up the understanding and design of heterogeneous catalysts in order to create more environmentally friendly processes. GAME-Net is open to exciting new additions like more complex catalyst materials and specific reaction steps.
“Our model demonstrates that AIs are capable of describing complex chemical interactions using extremely simple models, making them more accessible to scientists who want to accelerate their research,” conclude Dr. Pablo-Garca and Santiago Morandi. “Beyond the short-term impact our work may have,” they add,
More information: Sergio Pablo-García et al, Fast evaluation of the adsorption energy of organic molecules on metals via graph neural networks, Nature Computational Science (2023). DOI: 10.1038/s43588-023-00437-y




