Endogenous factors like reactive oxygen species and environmental factors like ultraviolet light, radiation, and chemicals constantly damage our genomic DNA. Inability to fix damaged DNA might prompt transformations and cell demise, at last prompting the beginning of malignant growth and different sicknesses. To forestall this, our phones are outfitted with different guard frameworks geared toward finding and fixing damaged DNA.
For the purpose of repairing a variety of DNA lesions brought on by chemical carcinogens and ultraviolet light, nucleotide excision repair (NER) is an essential mechanism. Numerous patients determined to have xeroderma pigmentosum (XP) have transformations in one of the qualities encoding proteins engaged with the NER system. They are known to be particularly susceptible to skin cancer caused by ultraviolet light because they lack NER function. Additionally, this indicates that NER protects against cancer.
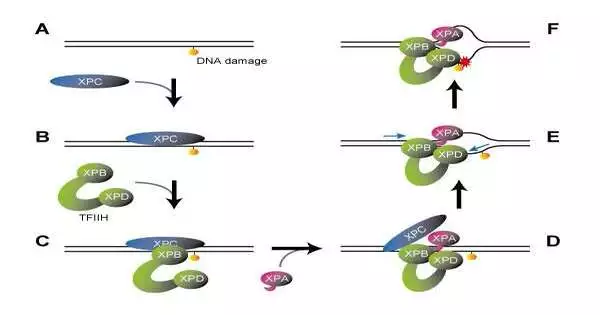
One of the many XP-causative gene products, the XPC protein, serves as a “sensor” to locate DNA structure anomalies in the first step of NER. In addition to binding to DNA sites without causing damage, such as base mismatches, XPC has the potential to deal with a wide range of DNA lesions. It is essential to check to see if there is any damage that needs to be fixed because unneeded repair reactions at these inappropriate locations may raise the likelihood of undesirable mutations. Although it has previously been demonstrated that the XPA protein and the basal transcription factor TFIIH are involved in this damage verification step, the specific mechanism has remained a mystery up until this point.
In a study that was published in Nature, the researchers prepared a series of complexes containing damaged DNA that were sequentially bound by XPC, TFIIH, and XPA. These complexes were then subjected to cryogenic electron microscopy for a detailed analysis of their molecular structures. The XPB and XPD proteins are two of the seven subunits that make up the large TFIIH protein complex, which is essential for both NER and transcription (gene expression). The XPB and XPD proteins are positioned at the tips of its two open arms, giving it a typical “horseshoe” shape.
The DNA-XPC-TFIIH complex maintained this horseshoe structure, with XPB bound to XPC at the damage site and XPD not interacting with either DNA or XPC. TFIIH, on the other hand, underwent a significant conformational change when XPA was added later. While the two open arms of the horseshoe met up to frame a ring-molded structure, XPB created some distance from the harmed site to such an extent that XPD was permitted to tie in the middle between.
Then, how does this molecular arrangement check for DNA damage? It is referred to by those XPB capabilities as a “DNA translocase,” which ties to the DNA duplex and moves along one of the DNA strands. It was assumed that XPB should move away from the damage site along the DNA based on the complex’s newly revealed structure. However, XPB cannot actually move because it is restricted by other proteins. All things being equal, the DNA duplex moves the other way. To put it another way, XPB introduces DNA into the complex.
It was anticipated that the structure would unwind the DNA duplex into single strands like a fastener being unzipped because it revealed that a portion of XPA is inserted between the two DNA strands behind XPB. In addition, it was previously understood that XPD moves along single-stranded DNA in a predetermined direction (5′, 3′).
Of the two DNA strands unwound by XPB and XPA, XPD ties to the damaged one. However, the damaged strand is also pulled toward the complex because the XPD itself cannot move, just like the XPB. XPD forms a small pinhole that allows the DNA strand to pass through when bound to single-stranded DNA. As a result, a bulky lesion on the DNA strand will be trapped at the XPD entrance, preventing the strand from moving (much like a knotted thread cannot pass through a needle’s eye).
The current examination in this way effectively explained the component by which the presence of a DNA sore is checked: The XPD blockage of DNA strand movement determines whether the repair reaction will proceed. In fact, previous studies have shown that for non-bulky DNA lesions (base detachment, for example), the NER repair process is not applicable to objects that could be anticipated to pass through the XPD hole.
The current study has made it clear which proteins, like XPA, XPB, and XPD, are involved in the NER process for repairing damaged DNA and how they work. In XP patients, structural changes induced in pathogenic variants of these proteins have a negative effect on the repair response. As a result, it may be possible to support the development of drugs and other treatments in the future.
More information: Jinseok Kim et al, Lesion recognition by XPC, TFIIH and XPA in DNA excision repair, Nature (2023). DOI: 10.1038/s41586-023-05959-z





{kind=link}