Impeding the restraint of PKD1 and PKD2 quality articulation by erasing a limiting site for microRNAs upsets the development and development of kidney sores in autosomal prevailing polycystic kidney illness (ADPKD) models, UT Southwestern scientists detailed. The discoveries, published in Nature Correspondences, propose a system for quality treatment with the possibility to capture or fix ADPKD.
For over 25 years, we have realized that ADPKD is brought about by changes in PKD1 or PKD2 qualities. However, no helpful system exists to pursue these main drivers, “said Vishal Patel, M.D., academic partner of Inner Medication in the Division of Nephrology at UTSW and the creator of the paper.”
ADPKD is among the most well-known human hereditary conditions and the most regular hereditary reason for kidney disappointment, influencing an expected 12.5 million individuals around the world. ADPKD is an acquired illness where patients commonly acquire one changed copy of PKD1 (or PKD2) and one typical duplicate.
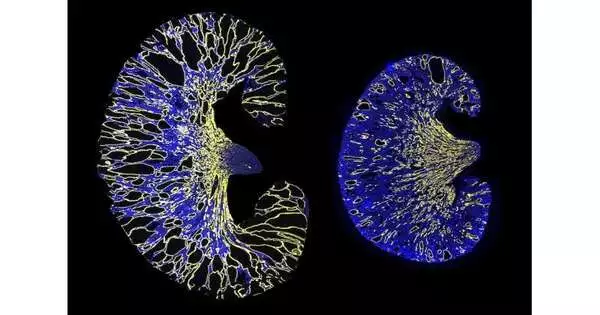
The illness is described by the regular development of numerous little liquid-filled sacs called kidney sores, which are considered to frame when the degrees of PKD1 or PKD2 fall under a basic edge. This can happen when the typical duplicate of the quality doesn’t create enough of the proteins Polycystin-1 and Polycystin-2.
Proteins are created (or deciphered) from a quality’s courier ribonucleic acid (mRNA). A region of code on one side of the mRNA strand protects it from corruption while also controlling the amount of protein produced.The limiting of microRNAs to this area of the mRNA code can hinder interpretation, prompting the creation of fewer proteins.
“There are many genetic diseases in which one copy of the causal gene is mutated but the other copy is normal. Our method for utilizing the remaining normal copy is likely to be relevant to many other disorders besides PKD.”
Vishal Patel, M.D., Associate Professor of Internal Medicine in the Division of Nephrology at UTSW
PKD1 contains a limiting site for miR-17, a microRNA that is profoundly communicated and dynamic in models of ADPKD. Thus, Dr. Patel and his partners inquired as to whether impeding the limiting of miR-17 to PKD1 could forestall kidney sore arrangement.
The scientists erased the miR-17 restricting site from PKD1 mRNA in cell societies and an ADPKD mouse model. Their outcomes showed that erasure of the limiting site expanded security of the mRNA strand, raised Polycystin-1 levels, and diminished kidney sore development. Also, the group found that impeding miR-17 restricted to PKD1 mRNA with an enemy of miR-17 medication after sore development likewise diminished pimple development, showing that this connection could be a promising objective for polycystic kidney illness (PKD) treatment.
“There are various hereditary circumstances where one duplicate of the causative quality is changed, yet the other duplicate is as yet typical. “Our way of dealing with tackling the excess typical duplicate is logical material for numerous different illnesses other than PKD,” said Dr. Patel.
More information: Ronak Lakhia et al, PKD1 and PKD2 mRNA cis-inhibition drives polycystic kidney disease progression, Nature Communications (2022). DOI: 10.1038/s41467-022-32543-2
Journal information: Nature Communications





{kind=link}