A global group of scientists has fostered a strategy for changing one class of anti-toxins by utilizing tiny creatures that produce these mixtures normally.
The discoveries, published July 25 in Nature Chemistry, could prompt the more proficient creation of anti-toxins that are viable against drug-safe microbes.
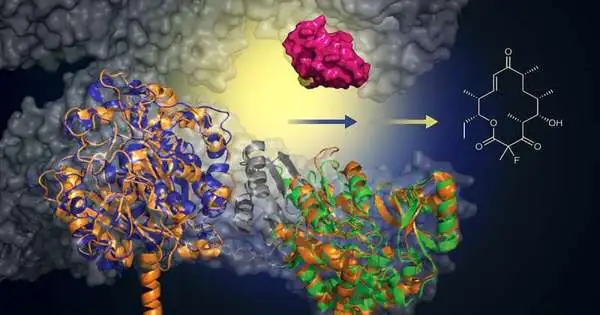
The group began with a microorganism that is hereditarily modified to create the anti-toxin erythromycin. Researchers from the Institute of Organic Chemistry and Chemical
Science at Germany’s Goethe University contemplated whether the framework could be hereditarily changed to gather the anti-toxin with one extra fluorine iota, which can frequently work on drug properties.
“We had been studying fatty acid synthesis for several years when we discovered a component of a mouse protein that we thought might be utilized for directed biosynthesis of these modified antibiotics if given to a biological system that can already manufacture the native molecule,”
Martin Grininger, professor for biomolecular chemistry at Goethe University.
“We have been examining unsaturated fat blends for a long time when we recognized a piece of a mouse protein that we accepted could be utilized for coordinated biosynthesis of these changed anti-toxins, whenever added to a natural framework that can as of now make the local compound,” said Martin Grininger, teacher for biomolecular science at Goethe University.
Working with the lab of David Sherman at the University of Michigan, which has some expertise in this organic gathering framework, the group utilized protein design to supplant one piece of the framework’s local hardware with a practically comparable mouse quality.
“It resembles removing one motor part from a Mercedes and placing it into a Porsche to make a superior mixed motor.” You get a Porsche motor that can do new things and works far better, “said Sherman, an employee at the U-M Life Sciences Institute and teacher of restorative science in the College of Pharmacy.
“We can now exploit this protein design to make new mixtures that have this truly helpful fluorine iota, which scientists have been battling to add to macrolide anti-toxins for quite a while.”
The reason this additional fluorine iota is so alluring is that it changes the design of the end result while maintaining the item’s capacity to kill microbes and work securely in patients.
Erythromycin works by restricting and impeding the action of the bacterial ribosome, which is fundamental for microbes to get by. A few microbes have developed ways of forestalling this limitation, making them impervious to treatment with anti-toxins. Changing the anti-toxin’s design with a fluorine iota beats that developmental benefit, reestablishing the compound’s capacity to battle microbes.
While physicists have created strategies for adding fluorine artificially, the cycle is strenuous and requires the utilization of harmful compound reagents. The new biosynthetic strategy created by the analysts from Goethe University and U-M beats those difficulties.
“It’s an extremely thrilling turn of events, since we can sidestep constantly consuming engineered advances and risky synthetics,” Sherman said. “We have demonstrated the way that we can essentially reinvent a creature to make the fluorinated item straightforwardly.”
The researchers emphasize that fluorinated compounds are still a couple of years away from being available in the market. Yet, the discoveries offer a more effective way ahead for growing new anti-toxins and even antivirals and hostile-to-disease meds.
“Our methodology has been demonstrated fruitful on a small number of anti-toxins, yet it could eventually be used to develop many drugs with negligible use of harmful synthetics,” Grininger said.
More information: Alexander Rittner et al, Chemoenzymatic synthesis of fluorinated polyketides, Nature Chemistry (2022). DOI: 10.1038/s41557-022-00996-z
Journal information: Nature Chemistry




