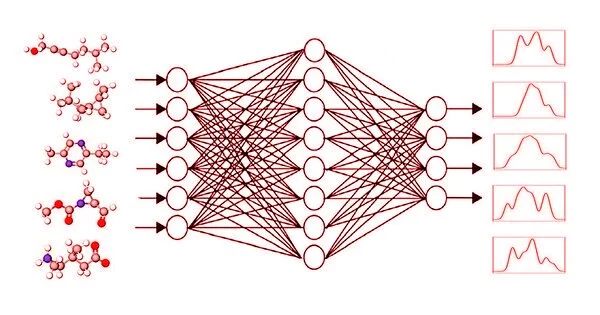
Calculating the spectral fingerprint of larger molecules using traditional methods is extremely time-consuming. However, this is required in order to correctly interpret experimentally obtained data. Now, using self-learning graphical neural networks, a team at HZB has achieved very good results in significantly less time.
“Macromolecules, but also quantum dots, which often consist of thousands of atoms, are difficult to calculate in advance using conventional methods like DFT,” says PD Dr. Annika Bande of HZB. She and her colleagues are now investigating how artificial intelligence methods can be used to reduce computing time.
The concept is as follows: a computer program from the “graphical neural networks” (GNN) group receives small molecules as input and is tasked with determining their spectral responses. The GNN program then compares the calculated spectra to the known target spectra (DFT or experimental) and corrects the calculation path as necessary. The outcome improves round after round. As a result, the GNN program learns on its own how to calculate spectra reliably using known spectra.
We trained five newer GNNs and discovered that one of them, the SchNet model, can achieve enormous improvements: the accuracy increases by 20% in a fraction of the computation time.
Kanishka Singh
“We trained five newer GNNs and discovered that one of them, the SchNet model, can achieve enormous improvements: the accuracy increases by 20% in a fraction of the computation time,” says first author Kanishka Singh. Singh is a student at the HEIBRiDS graduate school, where he is supervised by two experts from different fields: computer science expert Prof. Ulf Leser from Humboldt University Berlin and theoretical chemist Annika Bande.
“Recently developed GNN frameworks could do even better,” she claims. “And the demand is very high.” As a result, we want to strengthen this line of research and plan to create a new postdoctoral position for it starting this summer as part of the Helmholtz project “eXplainable Artificial Intelligence for X-ray Absorption Spectroscopy.”
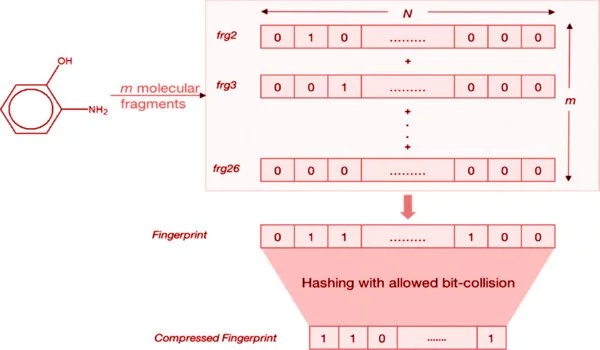
Fingerprints or descriptors are abstract representations of a molecule’s structural features. These descriptors could be structural keys within a molecule. This could be as simple as a count of a specific atom type, such as S, N, or halogen, or sp3. It could be the presence of a specific ring system, such as Phenyl, Pyridyl, or Naphthyl, or a functional group, such as Amide, Ester, or Amine. It could be a calculated property such as Hydrogen Bond donor, Polar Surface area, or LogP. Fingerprints are more abstract than structural keys, but they are more general because they do not represent pre-defined patterns.
Fingerprinting based on paths FP2, a path-based fingerprint that identifies small-molecule fragments based on linear segments of up to seven atoms. The structure of a molecule is examined in order to identify linear fragments ranging in length from 1 to 7 atoms. Single-atom C, N, and O fragments are ignored. When the atoms form a ring, the fragment is completed.
For each of these fragments, the atoms, bonding, and whether or not they form a complete ring are recorded and saved in a set, so that each fragment type exists only once. Chemically identical versions (atoms listed in reverse order and rings starting at different atoms) are identified, and only one canonical fragment is retained. Each remaining fragment is assigned a hash number from 0 to 1020 which is used to set a bit in a 1024 bit vector.


{kind=link}