An organic catalyst that performs better than known catalysts was discovered by a group of researchers using a computational approach inspired by evolution. A genetic algorithm suggested new catalytically active molecular structures for a common reaction in organic synthesis, as the team reports in the journal Angewandte Chemie, International Edition. The team says that the method could be used more broadly to find better molecular catalysts.
In a wide range of chemistry fields, machine learning systems are already able to predict material properties and molecular structures with high precision. In any case, robotizing the quest for better than ever impetuses has, to date, not been imaginable, although growing new impetuses for synthetic responses is quite possibly the main objective in substance research. With catalysts that work better, it’s possible to have reactions happen faster, easier, and with fewer byproducts and less energy.
The purpose for the troubles experienced via computerized frameworks while looking for new impetuses lies in response change states, as Jan Halborg Jensen, Teacher of Computational Science at the College of Copenhagen, Denmark, and relating creator for the review, makes sense of. This is because the transition state is influenced by catalysts; essentially, it is the point in a reaction that determines whether or not a product will be formed. Models are challenging to construct because of this moment’s fleeting nature and the intricate structures that result from the simultaneous interactions of numerous molecules.
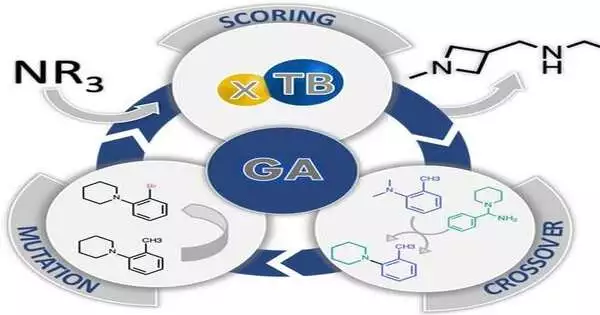
“The fittest molecules are then mated, which means that the two parents are cut in random locations and pieces from each parent are recombined. If you repeat this process several times, the ultimate population will look substantially different from the beginning population, much like a chihuahua varies from its wolf forebears.”
Jan Halborg Jensen, Professor of computational chemistry at the University of Copenhagen, Denmark.
Jensen and the team used an evolutionary selection strategy to overcome this obstacle. A set of starting molecules was evaluated using a genetic algorithm to see if they were suitable for catalyzing the Morita–Baylis–Hillman (MBH) reaction. After that, “you take the fittest molecules and mate them,” which entails randomly cutting the two parents and recombining fragments from each parent, as explained by Jensen. Similar to how a chihuahua differs from its wolf ancestors, the final population can look very different from the initial population if it is done repeatedly.
The four-membered azetidine ring, a novel structural motif absent from the initial population, was present in the computer-generated final molecules in this manner. The team then synthesized and tested one of the computer-evolved azetidine candidates in the reaction, and it outperformed the conventional catalyst, DABCO (1,4-diazabicyclo[2.2.2]octane). “The algorithm made a truly novel discovery because azetidines had never been considered as catalysts for the MBH reaction,” says Jensen, highlighting the significance of computer-assisted discoveries in chemical research.
Jensen says that a fundamental element for the utilization of this method is the information on the key change state for the response being referred to. He thinks that if this is known, genetic algorithms could help find new organ catalysts that are better.
More information: Julius Seumer et al, Computational Evolution Of New Catalysts For The Morita–Baylis–Hillman Reaction**, Angewandte Chemie International Edition (2023). DOI: 10.1002/anie.202218565
The data and code are available at sid.erda.dk/sharelink/hGBkdGdCy7 and github.com/jensengroup/mbh_catalyst_ga respectively.




