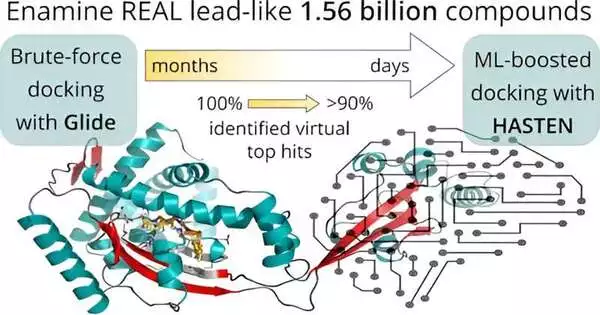
Helping with virtual screening with AI considered a 10-overlap time decrease in the handling of 1.56 billion medication-like particles. Scientists from the College of Eastern Finland collaborated with industry and supercomputers to create one of the world’s biggest virtual medication screens.
In their endeavors to find novel medication particles, specialists frequently depend on quick PC-aided screening of huge compound libraries to recognize specialists that can hinder a medication target. Such an objective can, for example, be a catalyst that empowers a bacterium to endure anti-infection agents or an infection to contaminate its host. The size of these assortments of little natural particles has seen an enormous flood over the course of the last few years.
With libraries becoming quicker than the speed of the PCs expected to handle them, the screening of a cutting-edge billion-scale compound library against just a solitary medication target can require a while or years—in any event, while utilizing cutting-edge supercomputers. Accordingly, obviously, quicker approaches are frantically required.
In a review distributed in the Diary of Substance Data and Displaying, Dr. Ina Pöhner and partners from the College of Eastern Finland’s School of Drug Store collaborated with the host association of Finland’s strong supercomputers, CSC—IT Place for Science Ltd.—and modern colleagues from Orion Pharma to concentrate on the possibility of AI in the acceleration of giga-scale virtual screens.
“This project is an excellent example of academic-industry collaboration, as well as how CSC can provide one of the best computational resources in the world. It was possible to achieve our lofty aims by combining our ideas, resources, and technology.”
Professor Antti Poso, who leads the computational drug discovery group within the University of Eastern Finland’s DrugTech Research Community.
Prior to applying man-made reasoning to speed up the screening, the scientists originally settled on a benchmark: In a virtual screening effort of extraordinary size, 1.56 billion medication-like particles were considered in contrast to two pharmacologically pertinent focuses over nearly a half year with the assistance of the supercomputers Mahti and Puhti and sub-atomic docking. Docking is a computational method that squeezes the little particles into a limiting district of the objective and figures out a “docking score” to communicate how well they fit. Along these lines, docking scores are not set in stone for all 1.56 billion atoms.
Then, the outcomes were contrasted with an AI-aided screen utilizing Rush, an instrument created by Dr. Tuomo Kalliokoski from Orion Pharma, a co-creator of the review.
“Rush purposes AI to get familiar with the properties of particles and what those properties mean for how well the mixtures score. When given an adequate number of models drawn from traditional docking, the AI model can foresee docking scores for different mixtures in the library a lot quicker than the savage power docking approach,” Kalliokoski makes sense of.
For sure, with just 1% of the entire library docked and utilized as preparing information, the device accurately recognized 90% of the best-scoring compounds within 10 days.
The review addressed the primary, thorough examination of an AI-supported docking device with a traditional docking benchmark on the gigascale. “We found the AI helped the device to dependably and more than once replicate most of the top-scoring intensities distinguished by customary docking in a fundamentally abbreviated time period,” Pöhner says.
“This undertaking is an amazing illustration of joint effort among the scholarly community and industry and how CSC can offer perhaps the best computational asset on the planet. By joining our thoughts, assets, and innovation, it was feasible to arrive at our aggressive objectives,” said Teacher Antti Poso, who drives the computational medication disclosure bunch inside the College of Eastern Finland’s DrugTech Exploration People group.
Concentrates on a tantamount scale stay tricky in many settings. Accordingly, the creators delivered enormous datasets produced as a component of the review into the public domain. Their preparedness to involve evaluating libraries for docking empowers others to accelerate their separate screening endeavors, with 1.56 billion compound-docking results for two focuses that can be utilized as benchmarking information.
This information will energize the future improvement of devices to save time and assets and will at last propel the field of computational medication revelation.
More information: Toni Sivula et al, Machine Learning-Boosted Docking Enables the Efficient Structure-Based Virtual Screening of Giga-Scale Enumerated Chemical Libraries, Journal of Chemical Information and Modeling (2023). DOI: 10.1021/acs.jcim.3c01239




