Scientists from numerous organizations in China have figured out how to utilize quality altering to reactivate lethargic fetal oxygen-shipping proteins in grown-up platelets to possibly switch an extensive variety of blood issues.
In a paper, “Base altering of the HBG advertiser prompts strong fetal hemoglobin articulation with no distinguishable off-target transformations in human HSCs,” distributed in Cell Foundational Microorganism, the group looks at quality altering strategies while planning a strategy that could have significant clinical applications.
Fetal gamma (γ) globin is regularly supplanted by grown-up (β) hemoglobin during advancement. In an odd peculiarity of development, just people and a couple of sorts of monkeys are known to change from γ to β quality articulation.
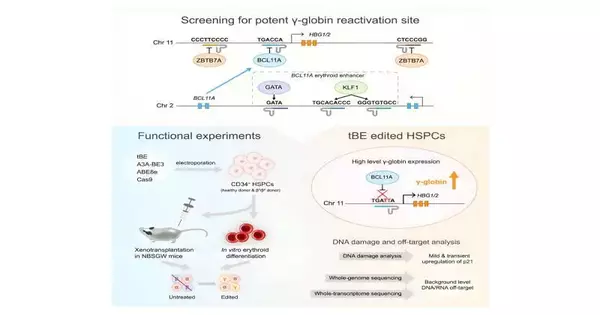
The qualities delivering fetal hemoglobin become hushed and torpid after the hereditary switch by repressors, for example, BCL11A and ZBTB7A, whose limiting themes have been distinguished as focuses for reactivation.
β-hemoglobinopathies, including β-thalassemia and sickle cell illness, result from transformations in the HBB quality, prompting weakened β-globin creation and bringing about iron deficiency, impeded oxygen conveyance to tissues, and conceivable multi-organ tissue harm.
The scientists tentatively found that reactivating γ-globin articulation could be formed into an all-inclusive helpful system for these circumstances.
Six administrative themes (BCL11A enhancer and HBG1/2 advertiser districts) were designated utilizing an as-of-late evolved cytosine base manager—the transformer base proofreader (tBE). The group contrasted tBE, other base editors, and Cas9 nuclease for productivity and off-target impacts.
In the review, tBE showed similar or higher altering effectiveness than different editors across the designated themes. Exhaustive examination uncovered no recognizable off-target changes in tBE-altered cells, showing the capability of tBE as a more secure and powerful treatment procedure for β-hemoglobinopathies.
Tests directed at patient-inferred cells revealed that upsetting the BCL11A restricting destinations inside the HBG1/2 advertisers prompted the most significant levels of γ-globin articulation. Xenotransplantation in mice showed relentless alteration in HSCs and their descendants, keeping up with engraftment potential and separation capacity.
The expanded γ-globin articulation seen because of tBE-interceded altering means a promising and helpful road for β-hemoglobinopathies.
While altering techniques and not immediate clinical results were the focal point of the review, the significant upgrade in γ-globin articulation levels firmly recommends possible clinical advantages, including side effect easing and further developed sickness for people impacted by β-hemoglobinopathies.
More information: Wenyan Han et al. Base editing of the HBG promoter induces potent fetal hemoglobin expression with no detectable off-target mutations in human HSCs, Cell Stem Cell (2023). DOI: 10.1016/j.stem.2023.10.007





{kind=link}