Stanford College scientists have fostered a computational strategy for distinguishing where cells are arranged in an example while capturing spatial transcriptomics. The technique uses information from spatial records and a reference single-cell RNA chart book to demonstrate yields. The subsequent models can be utilized to see cell bases, distinguish colocalization designs, and investigate differential articulation inside a cell type by area.
The new strategy — cell spatial situating examination through forced articulation (CytoSPACE)—has been published in the journal Nature Biotechnology. The content needed to run the calculation has been made openly accessible through GitHub.
Clinical exploration is exceptionally keen on understanding connections inside and between cells. While the genome has shown us a great deal about hereditary connections to sickness, the outflow of qualities in various tissues fluctuates generally. The job of limited cell tissues in foretelling or showing sickness is basic to understanding, foreseeing, diagnosing, and treating a huge scope of illnesses.
“We show that CytoSPACE outperforms earlier approaches in terms of noise tolerance and accuracy across many platforms and tissue types, enabling tissue mapping at single-cell resolution.”
The current study research group.
Cells inside tissues speak with one another, sending group or individual compound messages, continuously fussing over the solid situation of cell capabilities. On account of malignant growth, it is in many cases a breakdown in correspondence from inside a solitary cell that can prompt the illness to shape as tumorous cells lose the capacity to pay attention to the gathering’s guidelines and quit developing and fall to pieces.
Single-cell RNA sequencing can catch quality articulation (RNA particles) in individual cells with a high goal considering correlation between different cells. This strategy begins with disengaging single cells from the tissue of interest. While it teaches a scientist everything concerning communicated qualities inside the singular cell, it doesn’t express anything about the cells around it.
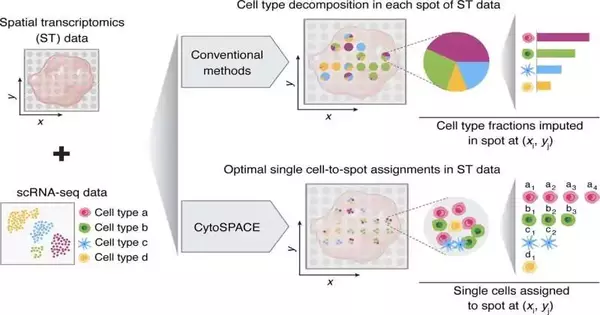
Current strategies for spatial transcriptomics take a more geological outline of quality articulation to make a guide of cell connections. Rather than individual cell data, scientists get a test of at least ten cells added to an area. Basically, a tissue test is put against an RNA catch slide that connects standardized tags to the RNA. At the point when the RNA is eliminated from the slide and sequenced, the standardized identification can be followed back to its assortment area on the slide, and thusly, the spatial game plan of the sequenced peruses can be reproduced into a guide.
In the quest for additional and exact information, a few computational techniques have been created to derive general cell structure from spatial transcriptomic tests. CytoSPACE uses single-cell information from a reference scRNA-seq map book and positions it at a higher level.
CytoSPACE builds a putative arrangement of single-cell groupings matching what is anticipated to be available in the spatial transcriptomics test. It then infills those successions to produce a set of areas as per the anticipated cell thickness of each spatial read. With these matched sets, CytoSPACE takes on the tissue remaking task as a straight-task issue and ideally maps individual cells in scRNA-seq information to spatial directions.
The ebb and flow concentrated on by the exploration bunch that created CytoSPACE tried it against the few driving computational strategies accessible. As per the paper, “Across different stages and tissue types, we show that CytoSPACE beats past strategies regarding commotion resistance and exactness, empowering tissue map making at the single-cell level.”
More information: Milad R. Vahid et al, High-resolution alignment of single-cell and spatial transcriptomes with CytoSPACE, Nature Biotechnology (2023). DOI: 10.1038/s41587-023-01697-9




