Another strategy for dissecting protein gems — created by Cornell College scientists and given an out-of-control two-section name — could open up applications for new medication revelations and different areas of biotechnology and organic chemistry.
The development, which is described in a paper that was published on March 3 in Nature Communications, gives researchers the tools they need to interpret the data from X-ray crystallography experiments, which were once discarded because they were an essential method for studying the structures of proteins. A better comprehension of a protein’s movement, structure, and overall function may result from this work, which builds on a study published in 2020.
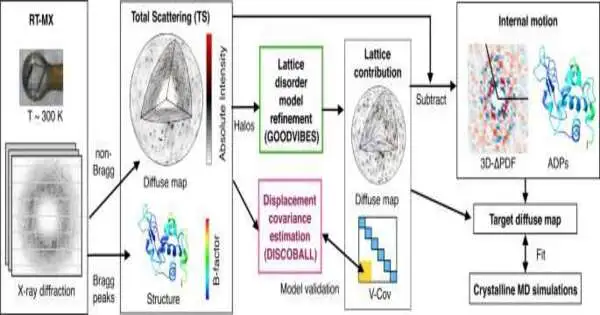
High-resolution information about a protein’s shape and structure can be obtained through protein crystallography, which produces bright spots from the crystals that are referred to as “Bragg peaks.” In addition, in the background of the Bragg peaks, this procedure captures blurry images—patterns and clouds related to the proteins’ movement and vibration.
“Analyzing it requires significantly more computing than just trying to evaluate crystallographic data alone. Diffuse scattering involves viewing everywhere at once, so we have to deal with a lot more data, and the signal is also highly complex.”
Nozomi Ando, associate professor of chemistry and chemical biology in the College of Arts and Sciences.
The bright Bragg peak imagery, which is easier to analyze, takes precedence over these background images.
Lead author Steve Meisburger, a former postdoctoral researcher in the lab of associate professor of chemistry and chemical biology Nozomi Ando, stated, “We know that this pattern is related to the motion of the atoms of the protein, but we haven’t been able to use that information.” We didn’t know how to use the information, even though it was there. Presently, we do.”
The robust diffuse scattering workflow, developed in close collaboration with Ando, is used to decode the weak background signals from crystallography experiments. The total scattering from crystals, which is influenced by the protein’s structure and the subtle blur of its movements, can now be studied by researchers thanks to this.
Their two-section strategy—which the group named GOODVIBES and DISCOBALL—at the same time gives a high-goal design of the protein and data on its corresponded nuclear developments.
By separating the protein’s subtle vibrations from those of other proteins that might be moving around it, GOODVIBES analyzes the X-ray data. For some proteins, DISCOBALL independently validates these movements directly from the data, allowing researchers to trust the GOODVIBES results and comprehend what the protein might be doing.
Ando expressed that while the potential for utilizing diffuse dispersing has been perceived from here onward, indefinitely, for quite a while, the demonstration of precisely estimating the inconspicuous sign while handling the information for something helpful has been undeniably challenging to do.
“It is substantially more computationally concentrated to examine than attempting to dissect crystallography information alone,” Ando said. “In diffuse scattering, we have a lot more data to deal with because we are looking everywhere at once, and the signal is also very nuanced.”
Ando expressed the general objective of transforming GOODVIBES and DISCOBALL into a certifiable underlying procedure that can be utilized by specialists at synchrotrons everywhere.
According to Ando, “there is a lot of interest within the fields of structural biology and biochemistry to use this signal.” We were not content with simply comprehending the signal’s contents; it was truly significant for us to make the following stage of making the apparatuses and making GOODVIBES and DISCOBALL accessible for others to utilize these instruments and test their speculations.”
These strategies were created utilizing lysozyme proteins gathered at the Cornell High Energy Synchrotron Source (CHESS). This spring, the Ando group will return to CHESS to work on more complex protein structures using their new method with Meisburger, who is now a CHESS staff scientist.
Researchers can gain a deeper understanding of how proteins move and interact with other significant molecules by separating the total scattering data of these complex proteins from the internal motion signals. This data can be utilized to configure new medications and treatments that target explicit proteins.
More information: Steve P. Meisburger et al, Robust total X-ray scattering workflow to study correlated motion of proteins in crystals, Nature Communications (2023). DOI: 10.1038/s41467-023-36734-3




